Latest News from the NCDIR
The NCDIR is actively expanding its outreach efforts to the general public!

The NCDIR director, Prof. Michael Rout will give a talk followed by a Q&A session at the Frontiers of Science Public Lecture Series hosted by Columbia University on November 27, 2023. He will explain how the NCDIR’s interactomic approaches have been applied to the study of a macromolecular assembly, the nuclear pore complex.
Event details:
Date: Monday, November 27, 2023
Time: 7:30 PM
Location: The Frank Altschul Auditorium, located at 417 International Affairs Building at Columbia University in New York City
(no registration required)
To learn more about the Frontiers of Science Program and the Public Lecture Series, visit https://ccnmtl.columbia.edu/projects/frontiers/
The NCDIR also places considerable emphasis on training young scientists of diverse backgrounds, leveraging multiple community outreach programs at our P41 performing sites, including the Summer Scholars Program (SSSP) at SCRI and the Summer Science Research Project (SSRP) hosted by RockEDU.
NCDIR Scientists present at the New York City Integrative Structural Biology Symposium
The NCDIR had the opportunity to disseminate its latest techniques and findings at the NYSBC (New York Structural Biology Center), in the New York City Integrative Structural Biology Symposium which was held on October 13, 2023, at the Advanced Science Research Center (ASRC) of CUNY. This interactive workshop was cohosted by the ASRC and NYSBC (New York Structural Biology Center) and open to the entire structural biology community. The focus of this symposium was to introduce integrative structural biology to our structural sciences community. The fields of light microscopy, mass spectrometry, X-ray crystallography, NMR, cryo-EM, and computational methods were highlighted.

Prof. Michael Rout presenting the keynote lecture
A group of scientists from the NCDIR presented their work at the event. Among them, Prof. Michael Rout was a keynote speaker. He gave a talk on “Setting the scene for integrative structural biology.” Prof. Brian Chait talked about “Mass spectrometry as a tool for examining dynamic macromolecular assemblies.” Dr. Trevor Van Eeuwen and Dr. Dom Olinares presented posters entitled “Structural analysis and inhibition of human LINE-1 ORF2 protein reveals novel adaptations and functions” and “Integrating native MS analysis into cryo-EM workflows applied to the characterization of SARS-CoV-2 replication-transcription assemblies.”
For more details about this workshop, visit https://nysbc.org/nyc-isb23/.
Research Spotlight — Implications of a multiscale structure of the yeast nuclear pore complex
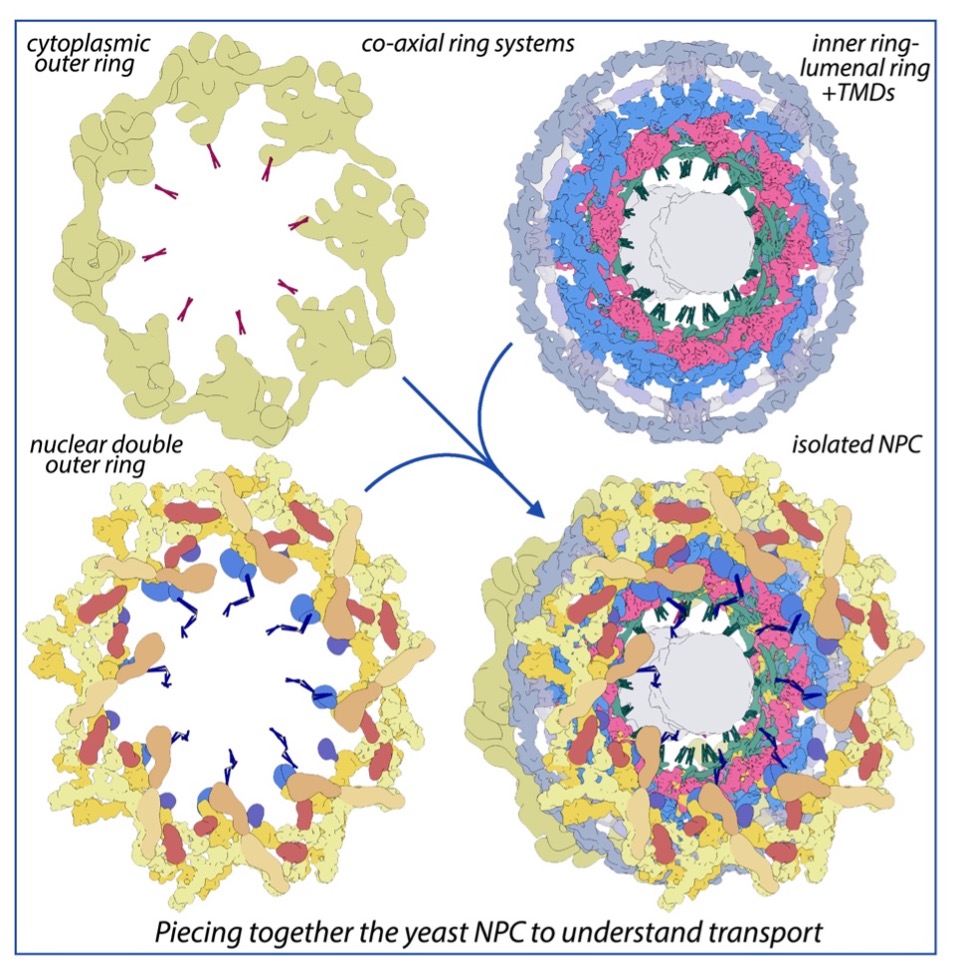
Nuclear pore complexes (NPCs) are the sole mediators of trafficking between the nucleus and cytoplasm. Sitting in the nuclear envelope, they are giant macromolecular assemblies that selective transport macromolecules bidirectionally to and from the nucleoplasm. In this latest update of our structural and functional mapping of the nuclear pore complex, we provide a composite multiscale structure of the yeast NPC, based on improved 3D density maps from cryogenic electron microscopy and artificial intelligence (AI) driven AlphaFold2 models of the individual nucleoporins comprising this structure. We resolved a host of flexible and dynamic connectors that tie together the entire core scaffold, providing a strong yet yielding architecture to the NPC. We have also begun to resolve the bundle of equatorial transmembrane regions that connect the NPC’s lumenal ring to the inner ring of the core scaffold, and so anchor the entire complex within the pore membrane. The organization of the nuclear double outer ring reveals an architecture that may be shared with ancestral NPCs. Additional connections between the core scaffold and the central transporter – the heart of the NPC’s transport machinery – suggest that under certain conditions, a degree of local organization is present at the periphery of this transport machinery. These connectors may couple conformational changes in the scaffold to the central transporter to modulate transport and suggest different regions of the central transporter may mediate distinct types of trafficking.
To read more, the paper can be freely accessed by clicking below:
You can subscribe to the latest NCDIR’s Rout lab news via twitter:
